1.1 实验材料
石油污染土壤S1,S2,S3,S4,S5,S6都取自陕西延安境内的油井,其中土壤S1和S2为短链为主的石油污染土壤(短链烃C11 - C20占60%),土壤S3,S4,S5为长链烃为主的石油污染土壤(长链烃C21 - C30占62%~64%),土壤S6未经石油污染(用于测定自由基).土壤试验前均经过碎散,去除杂质,用2 mm的细筛筛过后,在摇床上(125 rpm)转48 h混匀处理后密闭保存于冰箱(4 ℃)中待用混匀.五种土壤的理化性质见表1.
表1 5种土样样品的理化特性
Tab.1 Physical and chemical properties of five soil samples
1.2 化学药品
实验所用试剂主要包括:DMPO,5,5-二甲基-1-吡咯啉-N-氧化物(5,5-dimethyl-1-pyrroline N-oxide)和饱和烷烃标准品(C7-C30,99%)购买自美国Sigma-Aldrich公司,过氧化氢(H2O2,30% w/v,分析纯)从Paini试剂公司(郑州,中国)购买,柠檬酸(C6H8O7,分析纯)、硫酸亚铁(FeSO4·7H2O,分析纯)和无水氯化钙(CaCl2,分析纯)从National Medicine Group Chemical Reagent Factory(北京,中国)购买.二氯甲烷(CH2C12,HPLC级),氢氧化钠(NaOH,分析纯)和盐酸(HCl,分析纯),购自天津凯美化学试剂有限公司(中国天津).
1.3 实验步骤
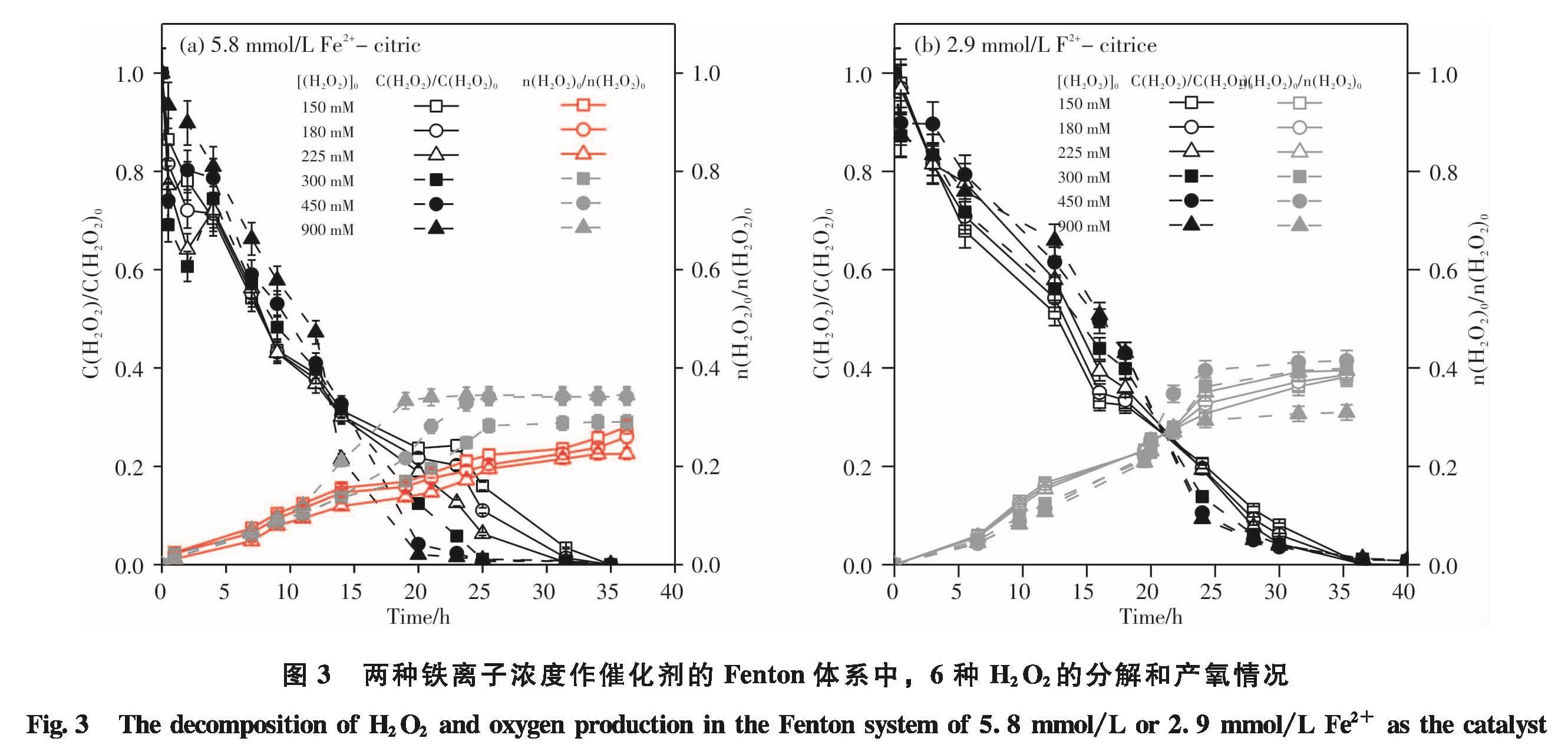
(1)低H2O2浓度体系中提高石油污染土壤的氧化效果的研究
为了探明低浓度H2O2投加时石油的降解情况,本实验设计了从高到低的6种H2O2投加浓度的Fenton氧化石油污染土壤的实验.
油氧化实验:设计在双氧水浓度从高到低6种浓度(900 mmol/L、450 mmol/L、300 mmol/L、225 mmol/L、180 mmol/L和150 mmol/L),高低两种Fe2+浓度(5.8 mmol/L和2.9 mmol/L)的条件下,对5种石油污染土壤(S1、S2、S3、S4和S5)进行油氧化实验.油氧化实验具体步骤如下:
取5g石油污染土样放入150 mL的锥形瓶,分别投加一定浓度的双氧水和Fe2+作为催化剂(柠檬酸作为螯合剂,浓度为15 mmol/L<sup>[15-16]和适量蒸馏水,保证体系最终体积为60 mL.在pH为7.0~7.5,室温(22±2 ℃)的条件下进行石油的氧化实验,待H2O2完全分解后测定体系中残留TPH和残留各碳链浓度.在相同实验条件下,做另外一组平行样,用来测定进入水相(non-aqueous phase liquid)的TPH(NAPL TPH)和各碳链浓度,研究减少双氧水浓度时对石油氧化与解析的影响.所有试验做了三份,每个样品测量三次.
(2)低浓度H2O2体系中提高石油氧化效果的机制研究
为了研究低浓度H2O2条件下石油氧化的机制.开展了H2O2分解实验、产氧实验和·OH测定实验三个部分.另外,Fenton实验条件同油氧化实验,土壤采用未污染土样S6,实验具体步骤如下:
H2O2分解实验:在反应期间定期测定体系中双氧水的残余浓度,待硫酸钛法显色后,使用UV-1240分光光度计测定体系中残余的H2O2浓度<sup>[17].
产氧实验:在反应期间定期用排水法收集体系中产生的氧气量,通过排出饱和的氢氧化钠溶液来收集氧气<sup>[18].
·OH测定实验:前期大量实验表明,·OH的产量与Fenton试剂关系密切,而土壤性质对其影响甚微.因此本实验选取具有代表性的未污染背景土样S6进行·OH产量的测定.采用电子自旋共振波谱仪(Bruker EPR A300)测定·OH的瞬时强度(记为It)随时间(记为t)的变化情况<sup>[19].羟基自由基测定的具体步骤如下:在摇匀后的泥水混合反应体系中取适量样品(大于20 μL),过0.22 μm的膜后取20 μL的溶液和10 μL100 mmol/L的捕获剂DMPO溶液,混合均匀,用毛细管吸取适量混合液后用橡皮泥堵住一端口后放入样品管,插入EPR的共振腔中进行测定.每次在投加H2O2后3 min之内取样测定·OH的瞬时强度,定期测定反应过程中·OH的瞬时强度随时间的变化规律,直至检测不到DMPO-OH信号.所有实验做了三份.最终积分·OH瞬时强度对时间的曲线,得到·OH产量,记为I=It*t.
此外再做三组空白实验,即在150 mL的锥形瓶中先加入5 g未污染土样S6,再分别加入以下溶液:(1)只加60 mL蒸馏水,(2)加适量蒸馏水和2.9 mmol/L或5.8 mmol/L的Fe2+(螯合剂为柠檬酸,浓度为15 mmol/L),(3)加适量蒸馏水和6种浓度的H2O2.使体系最终体积为60 mL,pH为7.0~7.5之间.发现三组空白实验中皆未检测到DMPO-OH信号峰.
1.4 分析方法
(1)·OH强度的测定
用布鲁克电子顺磁波谱仪(EPR)(Bruker EPR A300)在室温条件下用毛细管吸取长度约1 cm的混合液测定·OH强度,典型的几个EPR参数设置如下:中心磁场为3 507 G; 扫场宽度为100 G(用DMPO进行自旋捕获实验); 扫场时间为5.24 s; g因子为2.0; 接收获得为30 dB; 调制幅度为1.0 G; 调制频率为100 kHz; 扫描次数为5次; 微波衰减为20 dB<sup>[20-22].每组实验做3个平行样,每个样品测定三次.
(2)TPH萃取和分析
从石油污染土壤中萃取石油的过程按照US EPA test方法3550 B<sup>[23].向每个反应完成后的两个体系:a.水相和固相混合体系(为测定总残余TPH浓度)中、b.过滤掉上清液后(为测定除NAPL后吸附态TPH浓度)的固相体系中分别加入20 mL二氯甲烷,在摇床上摇24 h(125 rpm),再超声15 min后振荡30 min(150 rpm)使油从土壤中分离出来与二氯甲烷混合.用分液漏斗连续三次萃取油和二氯甲烷的混合物,并通过装有无水硫酸钠颗粒(提前在烘箱中105 ℃烘2 h)的滤纸去除水.最后在50 mL容量瓶中定容萃取的含油溶液至50 mL标线,测定a,b两个体系中的石油浓度.残余吸附态TPH浓度为b体系中萃取的石油浓度,非水相TPH的浓度为a体系中萃取的油浓度减去b体系中石油的浓度,氧化去除的TPH浓度为空白石油浓度减去a体系中的石油浓度<sup>[24].另外,我们计算了TPH和链烃的K值,具体计算方法是通过氧化率除以解析率得到的.
使用安捷伦6890N气相色谱仪(美国)分析测定样品; 检测器:FID检测器; 色谱柱:HP-5毛细管柱(30 m*0.25 mm*0.50 μm); 分流进样,分流比5:1; 进样量:1 μL; 进样口温度:300 ℃; 检测器温度:300 ℃; 载气:氮气,载气流速:30 mL/min,空气流速:300 mL/min,尾吹气流速:28 mL/min; 测样升温程序:三阶段升温程序,以40 ℃的柱箱温度保持0.5 min,再升温至150 ℃,保留2 min,速度为15 ℃/min,继续升温至290 ℃,速度为10 ℃/min,保留5 min,整个过程运行28.83 min.GC开始和结束时都用二氯甲烷和纯TPH混合标液进行测定.可用内部的标准(17烷)用于校准,从而获得一个正确的石油标准曲线(标准石油烃从Sigma 公司购买).TPH和各链烃的浓度可通过对比石油标准曲线计算出来.